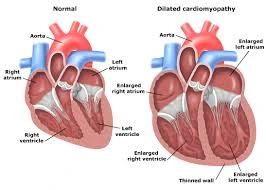
I ricercatori Chiara Mozzetta, dell’Istituto di biologia e patologia molecolari del Consiglio nazionale delle ricerche (Cnr-Ibpm, Roma) e Francesco Saverio Tedesco della University College London (UK) hanno pubblicato nel numero del 23 agosto della rivista Journal of Cell Biologyun loro commento su 'Nuove possibilità di comprensione e cura della cardiomiopatia dilatativa (Dcm) dalla riprogrammazione di cellule staminali'. La Dcm, chiamata anche ‘laminopatia’, è una patologia genetica delle cellule muscolari cardiache causata da mutazioni nel gene Lmna (che codifica per una proteina della lamina nucleare, Lamin A/C). La laminopatia cardiaca è caratterizzata dall’insorgenza precoce (tra i 30 e i 40 anni) di anomalie nella conduzione elettrica tra le cellule cardiache e/o dalla comparsa di aritmie maligne (es. fibrillazione atriale e tachicardia ventricolare) che possono portare a morte improvvisa. Tuttavia, non si conosce il meccanismo che collega le mutazioni in LMNA all’aumento di aritmogenicità. L’analisi parte da un recente studio del gruppo di Charles Murry dell’Università di Washington (Bertero et al., 2019; https://doi.org/10.1083/jcb.201902117), che ha utilizzato cardiomiociti, ottenuti da cellule staminali riprogrammate (induced-pluripotentstemcells, o iPSCs) da pazienti affetti da cardiomiopatia dilatativa. Sebbene sia noto da diversi anni che mutazioni nel gene LMNA causino patologie a carico del tessuto muscolare cardiaco e scheletrico, il meccanismo molecolare alla base di questi difetti è rimasto a lungo sconosciuto. La Lamin A/C fornisce supporto strutturale al nucleo delle cellule, in cui è contenuto il nostro genoma, e partecipa alla regolazione tri-dimensionale del genoma stesso. Quest’ultimo, associandosi a diverse proteine presenti nel nucleo delle cellule, si organizza in particolari strutture, i cosiddetti Topologically-Associated Domains (TADs). Da qui la necessità di capire se le mutazioni in Lmna, riscontrate nei pazienti, possano influire sulla regolazione dei TADs e sui difetti dell’espressione dei geni. La pubblicazione mette in evidenza le importanti novità rese possibili grazie all’uso dei cardiomiociti riprogrammati da pazienti affetti da DCM. Oltre a indurre difetti di espressione genica, riconducibili all’incapacità dei TADs di assemblarsi correttamente, l’assenza di una Lamin A/C funzionale potrebbe generare una aberrante espressione di geni, causa dei difetti clinici nelle proprietà elettriche dei cardiomiociti dei pazienti. La riprogrammazione delle cellule staminali e la progettazione di nuove terapie. Poiché evidentemente non sarebbe possibile prelevare cardiomiociti direttamente dai pazienti, il cardine fondamentale per queste scoperte e per altre rilevanti patologie è proprio nell’uso delle iPSCs, cellule somatiche che possono invece essere facilmente ottenute da pazienti con mutazioni specifiche in un particolare gene (in questo caso Lmna), e che possono essere indotte a differenziare in cellule cardiache in vitro. Questa strategia di ‘disease modelling’ si dimostra particolarmente efficace per identificare un legame specifico tra genotipo (la mutazione) e fenotipo (la manifestazione clinica) e per studiare i difetti “epigenetici”, legati all’espressione del genoma, associati alle diverse mutazioni. Durante il convegno annuale del Cnr-Ibpm (che si è tenuto lo scorso 9 maggio a Roma), Chiara Mozzetta, recentemente premiata da un finanziamento di Telethon France per i suoi studi sulle distrofie, ha discusso proprio dello sviluppo di queste strategie http://www.cnrweb.tv/ibpm-cnr-focus-su-malattie-neurologiche-e-e-oncologia/. La sessione sulle distrofie muscolari, è stata aperta da Giulio Cossu, autore del libro ‘La trama della vita’ e pioniere della riprogrammazione di cellule staminali di bambini affetti dalla distrofia muscolare di Duchenne. Il congresso ha inoltre presentato un approccio simile, sviluppato da Ferdinando Squitieri (associato CNR-IBPM, fondatore della Italian Research League for Huntington e direttore dell’Unità di Ricerca per la malattia di Huntington all’Istituto Mendel di Roma, IRCCS Casa Sollievo della Sofferenza) in collaborazione con Andrea Ilari (direttore della Bio Crystal Facility del CNR-IBPM, Roma), per la riprogrammazione di cellule staminali prelevate da pazienti con una rara forma giovanile della corea di Huntington. Questi nuovi approcci saranno fondamentali sia per capire i difetti molecolari alla base delle patologie, sia per la prospettiva, a lungo termine, di sviluppare nuove terapie cellulari basate sulla correzione genetica dei difetti e il trapianto delle cellule ‘corrette’.